https://www.asminternational.org/home/-/journal_content/56/10180/40278982/NEWS
Study indicates AlCe and AlCo aluminum binary alloys could be suitable for 3D-printing
April 20, 2020
Source: ASM International
Researchers from the University of Connecticut, Pratt & Whitney, and Collins Aerospace collaborated in an 18-month study in which they used computational tools and a variety of experimental methods to develop simple aluminum binary alloys with properties similar or superior to commercial 6xxx aluminum alloys for additive manufacturing.
The goal was to identify binary systems of the type Al-X that promise hardness advantages after processing under conditions found in a typical metal additive manufacturing environment. DFT and laser surface glazing experiments were used to identify alloy additions to aluminum that induce lattice strains around solute atoms, and that can yield extended solid solubilities.
Given the non-equilibrium nature of rapidly quenched alloys, solid solutions with extended solubilities are metastable in nature and might transform further, for example, into microstructures with clusters or second-phase precipitates. The current work therefore represents an approach to the design of new aluminum alloys specific to non-equilibrium process technologies such as additive manufacturing.
Results show conclusively that cerium and cobalt are promising elements in next-generation aluminum alloys that make use of non-equilibrium processing conditions such as additive manufacturing.
The investigation started with an analysis of solid solution strengthening using first-principles calculations to determine elastic property changes and local lattice distortions from the introduction of different elements into a host aluminum lattice. These results, coupled with both equilibrium and nonequilibrium solubility data, led to the selection of cerium and cobalt as the primary candidate alloying elements. Alloys of Al\\Ce and Al\\Co at concentrations of 0.5, 1.0, and 3.0 at. % were then synthesized and subjected to laser glazing to produce non-equilibrium microstructures.
The microstructure and solid solution characteristics were determined using a combination of scanning electron microscopy and transmission electron microscopy. Hardness was measured by nanoindentation testing, which showed that both candidate systems harden significantly after glazing. In addition, Al-1.0Co at. % achieves a hardness comparable to Al6061-T6.
Following are comments from distinguished materials scientists regarding this study:
Prof. Rainer Hebert, Director – Pratt & Whitney Additive Manufacturing Center, Department of Materials Science and Engineering, University of Connecticut
“This is a comprehensive study where we developed simple Al-binary alloys with similar (if not superior) properties compared to commercial 6xxx series Al alloys. We collaborated extensively with Collins Aerospace and Pratt & Whitney. As such, this is also a great example of industry and academia working side by side in addressing a fundamental problem in metallurgy.”
Prof. Diran Apelian, Distinguished Professor of MSE, Chief Strategy Officer- SSoE, UCI, Irvine, Calif.
“Beauty is usually used as an adjective describing objects, individuals, nature; I will use it here to a manuscript as it exemplifies excellence and a set of standards for us to aspire. A “beautiful” paper in that the hypotheses, the experiments, coupled with a deep dive in ICME calculations, and validation in collaboration with industrial partners, have been executed in a compelling manner. The results are most meaningful and impactful. Accolades to our UConn colleagues.”
Prof. Lesley Frame, Director – Center for Materials Processing Data (CMPD), an ASM International Consortium; Department of Materials Science and Engineering, University
“Successful studies that identify improved methods for materials discovery like this one, illustrate the need for reliable methods of characterizing transient properties of the novel materials. CMPD is taking on the challenge of generating and validating the materials processing data that will support rapid transition from materials discovery to manufacturing application.”
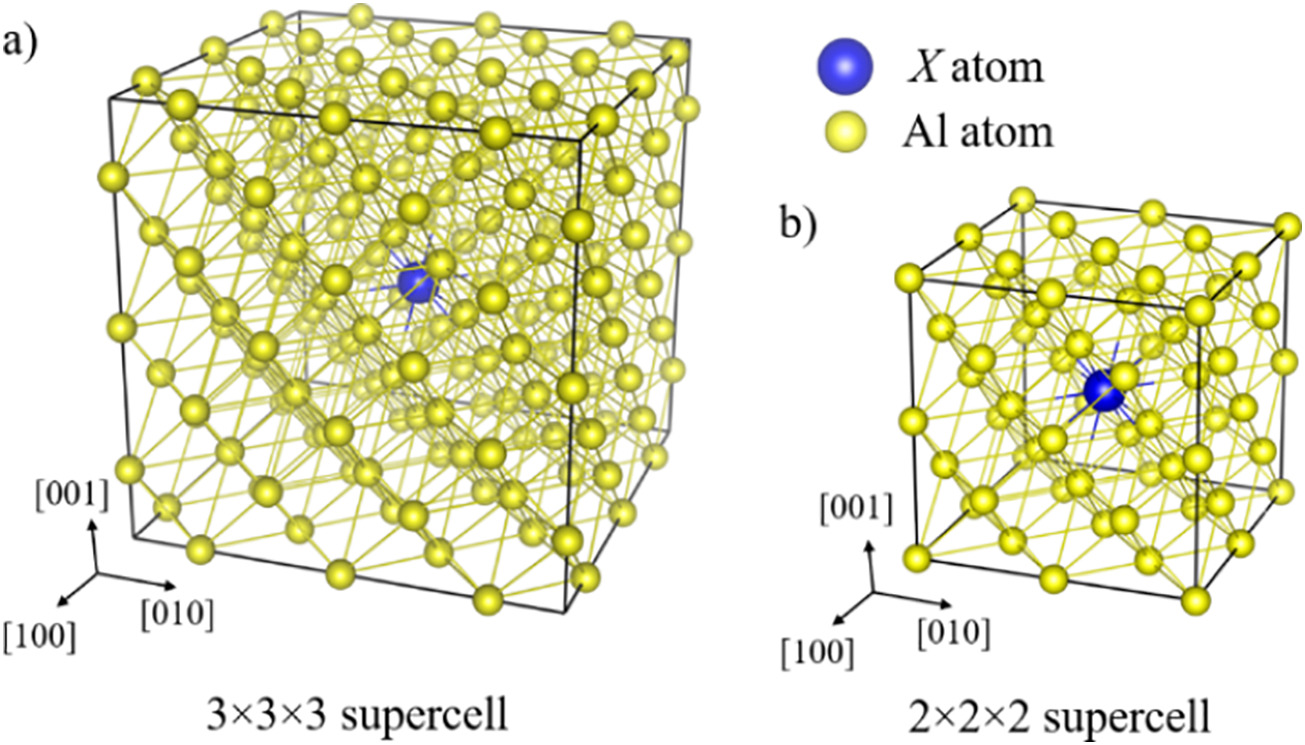
Image: Ball-stick models for supercells constructed from a) 3 × 3 × 3 unit cell and b) 2 × 2 × 2 unit cell, containing 108 atoms and 32 atoms, respectively. The aluminum atoms are represented as smaller yellow balls and the X atom, where X represents elements with atomic number 1–94 (H\\Pu) other than aluminum in the periodic table, is represented by larger blue ball. The ball sizes are not representation of atomic sizes. Substituting one X atom in the supercells leads to concentration of 0.926 at. % and 3.13 at. % for 108 atoms and 32 atoms supercells, respectively.
*************************
Full paper citation:
C. J Hung, S. K. Nayak, Y. Sun, C. Fennessy, V. K. Vedula, S. Tulyani, S.-W. Lee, S. P. Alpay, and R. J. Hebert, “Novel Al-X Alloys with Improved Hardness,” Materials & Design, 192, 108699 (2020); https://doi.org/10.1016/j.matdes.2020.108699